RESEARCH AREAS
The human genome is organized into 46 linear chromosomes whose copy number and sequence order have been precisely established over evolutionary timescales. Alterations and rearrangements to this linear sequence define structural chromosomal abnormalities – a pervasive genomic feature of human cancers and developmental disorders. Our laboratory aims to address important questions in cell biology and cancer genetics by employing an array of innovative approaches bridging genome engineering, high-resolution imaging of cellular dynamics, molecular cytogenetics, and genomics. We also seek to develop experimental systems to reconstruct the major events and outcomes of genomic instability and chromosomal rearrangements in human cells.
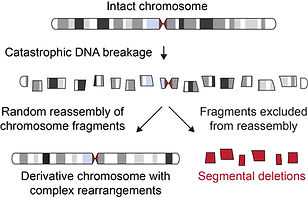
Chromothripsis and complex genomic aberrations
Cancer genome sequencing has uncovered novel classes of complex structural rearrangements that previously escaped recognition by classical cytogenetic approaches. This is epitomized by chromothripsis – large-scale genome rearrangements caused by the catastrophic shattering of an entire chromosome followed by its haphazard reassembly in random order. Chromothripsis can be initiated in mitosis by chromosome missegregation into aberrant cellular compartments called micronuclei. We are interested in understanding the mechanisms by which chromothripsis arises, as well as other simple and complex types of genomic rearrangements that are frequently associated with human disease, including chromosomal translocations, deletions, and insertions.


Metaphase spread illuminated by fluorescent DNA probes (green) recognizing a chromosome that missegregated into a micronucleus and shattered into multiple fragments.
Chromosome segregation errors driven by centromere dysfunction
Centromeres are specialized loci for kinetochore assembly, which facilitate chromosome movement through direct attachment to the mitotic spindle during cell division. Although human centromeres are typically located on repetitive alpha-satellite DNA sequences, these sequences are neither essential nor sufficient for centromere identity, maintenance, and function. Instead, human centromeres are defined epigenetically by the incorporation of the histone H3 variant 'centromere protein A' (CENP-A) into centromeric chromatin. By exploiting our knowledge of centromere biology along with rapid gene replacement technologies, we have developed experimental systems utilizing chimeric CENP-A mutants to selectively inactivate the centromere of a specific chromosome-of-interest – the human Y chromosome. This strategy has enabled us to reconstruct the mechanisms of chromothripsis, beginning with the induction of chromosome missegregation into micronuclei all the way toward producing fully functional and genetically stable chromothriptic rearrangements. We now seek to further leverage centromeres – including neocentromeres formed de novo at non-alpha-satellite DNA sequences – to study how centromere dysfunction contributes to genome instability.

DNA damage response and repair mechanisms
Chromosomes are constantly exposed to intrinsic and extrinsic sources of DNA damage, which can generate dangerous DNA lesions known as double-strand breaks (DSBs). Micronuclei have recently emerged as a potent source of genomic instability, leading to the accumulation of localized DNA breaks during interphase that resolve into shattered chromosome fragments in the subsequent mitosis. We previously identified that the spectrum of micronucleus-dependent structural chromosomal aberrations can be developed predominantly through a DSB repair mechanism called non-homologous end joining (NHEJ). Our lab is currently investigating (1) the causes of micronuclear DSBs across the cell cycle and (2) how classical and alternative NHEJ pathways spatially and temporally orchestrate the reassembly of broken chromosome fragments into producing complex genomic structural variation.

Consequences of chromosomal instability in human health and disease
By establishing experimental systems to reconstruct the complexities of the cancer genome, our laboratory aims to define the consequences of numerical and structural chromosomal abnormalities in driving cancer initiation and progression. To do so, we employ both non-transformed and transformed human cell models to interrogate how genomic rearrangements promote tumorigenesis. This could occur through DNA copy-number alterations that amplify oncogenes or delete tumor suppressor genes, as well as rearrangements that perturb 3D chromosome organization and architecture. Future work will center on identifying actionable therapeutic vulnerabilities associated with genomic instability.

DNA copy-number alterations from a clonal chromothripsis event induced by centromere inactivation. Colored lines represent individual rearrangement breakpoints resolved by whole-genome DNA sequencing.
